Bài viết mới nhất



Lysosome là gì? Chức năng tuyệt vời của Lysosome với cơ thể
Lysosome là khoang tiêu hóa chính của tế bào. Chúng chứa nhiều loại enzyme... suy giảm các dưỡng chất như: axit nucleic, lipid và protein ....Ở góc độ tế bào khái niệm Lysosome là gì cực kỳ quan trọng. Sau đây hãy cùng botchumngay.vn tìm hiểu về chức năng tuyệt vời của lysosome với cơ thể trong bài viết dưới đây nhé!
Lysosomes là gì?
Được ví như nhà máy xử lí rác của tế bào, Tiêu thể hay tên tiếng Anh là Lysosome chứa các enzyme thủy phân thực hiện chức năng phân hủy các sản phẩm thừa như protein, nucleic, acid, polysaccharide đảm bảo cho tế bào hoạt động ổn định. Ngoài ra Lysosome còn phân hủy cả những tế bào tổn thương, tế bào già hoặc chết.

Lysosome là một phần quan trọng trong tế bào sống con người
Cấu tạo hình thái
Kích thước, hình dạng của lysosome rất đa dạng và tuỳ thuộc vào các chất khác nhau mà thể lysosome thu thập vào để phân giải.
Lysosome là những khối hình cầu đường kính từ 0,2 - 0,4μ, có khi lớn đến 1 - 2μ. Lysosome được bao bởi một màng lipoproteide (màng tế bào).
Tuỳ thuộc vào sự hình thành, thành phần cũng như hoạt tính chức năng của các chất chứa trong lysosome mà người ta phân thành 4 dạng, trong đó chỉ có một dạng là nguyên phát còn 3 dạng kia là lysosome thứ phát.
Thể lysosome cấp I
Là dạng lysosome nguyên phát. Là khối hình cầu nhỏ, chứa những enzyme thuỷ phân. Những enzyme này sẽ hoạt động khi xung quanh trở thành acid (pH < 7).
Không bào tiêu hoá
Được tạo ra do sự gắn kết của không bào chứa dị vật với lysosome nguyên phát. Trong không bào tiêu hoá, dị vật dần dần bị phân huỷ nhờ sự hoạt động của các enzyme chứa trong lysosome nguyên phát.
Thể cặn bã
Khi các dị vật không bị phân huỷ hoàn toàn, những cặn bã còn tồn tại trong lysosome tạo thành thể cặn bã. Thể cặn bã sẽ bị tống ra khỏi tế bào.
Các không bào tự tiêu (còn được gọi là xitolysosome)
Là một dạng của lysosome chứa những cấu trúc của bản thân tế bào (ví dụ: các ty thể, ribosome, các mảnh của mạng lưới nội sinh chất,...) đang trong quá trình bị tiêu hoá. Vì vậy, nhiều tác giả gọi không bào tự tiêu là xitolysosome hoặc otolysosome. Không bào tự tiêu được hình thành trong các quá trình sinh lý hoặc bệnh lý.
Dựa vào quá trình tiêu hoá nội bào người ta còn phân biệt ra 3 kiểu lysosome như sau:
- Các yếu tố tiền lysosome, đó là các fagosome hay otofagosome.
- Các lysosome cấp I và cấp II (các fagolysosome, otolysosome hoặc xitolysosome).
- Các yếu tố hậu lysosome, tương đương với các thể còn lại.
- Các yếu tố tiền lysosome chỉ chứa các dị vật - là các đối tượng để phân giải mà không chứa enzyme.
- Các thể lysosome cấp I có chứa enzyme hydrolase-acid, nhưng chưa tham gia vào quá trình tiêu hoá.
- Các lysosome cấp II chứa các enzyme hydrolase, chứa cả các chất chưa bị tiêu hoá hoặc đang bị tiêu hoá.
Cấu tạo hoá học
Màng lysosom là màng sinh chất (màng tế bào) được cấu tạo từ protein và lipid. Hệ thống màng có nguồn gốc từ màng Golgi hoặc màng tế bào (Smith, 1969). Trong lysosom có chứa nhiều men thuỷ phân như: phosphatase acid, ADNase, ARNase, protease, lipase, glucosidase, collagenase, catepsin,... Hiện nay, người ta đã biết chính xác 40 loại men khác nhau có trong lysosom.
Những men này chỉ hoạt động ở trong môi trường acid (pH = 5) và chỉ được giải phóng ra khỏi lysosom bị phá huỷ.
Enzyme Lysosome
Lysosome chứa nhiều enzym thủy phân khác nhau (khoảng 50 enzym khác nhau) có khả năng tiêu hóa axit nucleic, polysaccharid, lipid và protein. Bên trong lysosome được giữ có tính axit vì các enzym bên trong hoạt động tốt nhất trong môi trường axit. Nếu tính toàn vẹn của lysosome bị tổn hại, các enzyme sẽ không gây hại nhiều trong tế bào trung tính của tế bào.

Bên trong lysosome được giữ có tính axit vì các enzym bên trong hoạt động tốt nhất trong môi trường axit
Sự hình thành Lysosome
Lysosome được hình thành từ sự hợp nhất của các túi từ phức hợp Golgi với các nội bào. Nội bào là những túi được hình thành bởi quá trình nội bào khi một phần của màng sinh chất bị chụm lại và được nội bào hóa. Trong quá trình này, vật chất ngoại bào được tế bào tiếp nhận. Khi các ống nội soi trưởng thành, chúng được gọi là ống nội soi muộn. Các ống nội tạng muộn hợp nhất với các túi vận chuyển từ Golgi có chứa các hydrolase axit. Sau khi hợp nhất, những nội tiêu này cuối cùng phát triển thành lysosome.

Lysosome được hình thành từ sự hợp nhất của các túi từ phức hợp Golgi với các nội bào
Lysosome sử dụng enzyme để thực hiện chức năng tiêu hóa nội bào, các chất cặn bã, dư thừa trong tế bào sẽ được Lysosome nhận diện, tiết enzyme hay còn gọi là men phù hợp để tiêu hóa. Các sản phẩm thừa sau qua trình tiêu hóa nội bào sẽ được chuyển hóa sử dụng lại hoặc thải ra ngoài.

Lysosome sử dụng enzyme để thực hiện chức năng tiêu hóa nội bào, các chất cặn bã, dư thừa trong tế bào
Đôi khi Lysosome còn tiêu hóa cả tế bào gọi là thực bào. Ấy là khi tế bào nhiễm bệnh, già, chết hoặc tổn thương không thể tiếp tục làm việc hiệu quả.
Sự tương đồng giữa Endosome và Lysosome
- Endosome và lysosome là hai loại túi liên kết màng bên trong tế bào.
- Cả hai đều chứa các chất quan trọng cho hoạt động của tế bào.
- Ngoài ra, cả hai có thể hình thành từ bộ máy Golgi.
- Hơn nữa, cả hai đều đóng một vai trò quan trọng trong endocytosis và phagocytosis.
Sự khác biệt giữa Endosome và Lysosome
Định nghĩa
Endosome đề cập đến một túi hình thành do sự xâm lấn và chèn ép của màng tế bào trong khi lysosome là một cơ quan trong tế bào chất của các tế bào nhân chuẩn có chứa các enzyme thoái hóa được bao bọc trong màng. Do đó, đây là sự khác biệt cơ bản giữa endosome và lysosome.
Sự hình thành
Hơn nữa, sự hình thành là một sự khác biệt lớn giữa endosome và lysosome. Endosome, chủ yếu, hình thành trong quá trình endocytosis trong khi lysosome hình thành từ bộ máy Golgi.
Các thành phần
Một sự khác biệt khác giữa endosome và lysosome là thành phần của chúng. Nội nhũ chứa các vật liệu nội hóa bao gồm các chất dinh dưỡng và mầm bệnh như vi khuẩn trong khi lysosome chứa các enzyme thủy phân.
Chức năng
Hơn nữa, endosome lưu trữ các vật liệu nội hóa cho đến khi tiêu hóa của chúng trong khi lysosome hợp nhất với endosome, hỗ trợ quá trình tiêu hóa vật liệu bên trong endosome. Vì vậy, đây là một sự khác biệt khác giữa endosome và lysosome.
Chức năng của lysosome
Các chức năng chính của lysosome là:
Tiêu hóa nội bào
Từ lysosome bắt nguồn từ "mịn" (lytic hoặc tiêu hóa) và "soma" (cơ thể). Các không bào pinocytic, được hình thành như là kết quả của sự hấp thụ chất lỏng trong không bào tế bào hoặc thực bào (được hình thành bởi sự hấp thụ các hạt rắn trong tế bào), vận chuyển vật liệu protein đến vùng lysosomal.
Những protein này có thể trải qua quá trình tiêu hóa trong tế bào do hậu quả của bệnh nội tiết. Endocytosis bao gồm các quá trình thực bào, pinocytosis và micropinocytosis.
Phagocytosis và pinocytosis là các cơ chế hoạt động trong đó tế bào cần năng lượng để hoạt động. Trong quá trình thực bào do tăng bạch cầu, tiêu thụ oxy, hấp thu glucose và phân hủy glycogen tăng đáng kể.
Trong endocytosis, sự co lại của các vi chất Actin và myosin có trong tế bào chất ngoại vi xảy ra. Điều này làm cho màng plasma xâm lấn và hình thành không bào nội tiết. Các hạt ăn vào được bao bọc trong màng có nguồn gốc từ màng plasma và hình thành không bào đôi khi là các phagơ tế bào.
Tiêu hóa nội bào là một trong những chức nắng chính của lysosome
Sau sự xâm nhập của một hạt lớn hoặc cơ thể vào trong tế bào bởi endocytosis và sự hình thành của một phagosome, màng của phagosome và lysosome có thể hợp nhất để tạo thành một không bào lớn duy nhất.
Trong không bào này, các enzyme lysosomal bắt đầu quá trình tiêu hóa vật chất lạ. Ban đầu lysosome, được gọi là lysosome chính, chứa phức hợp enzyme ở trạng thái không hoạt động, nhưng sau khi hợp nhất với phagosome, nó tạo ra một lysosome thứ cấp với một hình thái và enzyme hoạt động khác nhau.
Sau khi tiêu hóa enzyme, vật liệu được tiêu hóa khuếch tán vào màng tế bào của tế bào. Một số vật liệu có thể vẫn còn trong không bào của lysosome mở rộng. Không bào còn sót lại này là cơ thể còn lại, vì nó chứa dư lượng của quá trình tiêu hóa.
Trong quá trình chết đói, lysosome tiêu hóa các nguyên liệu thực phẩm được lưu trữ, tức là protein, lipid và glycogen từ tế bào chất và cung cấp năng lượng theo yêu cầu của tế bào. Quá trình tiêu hóa protein thường kết thúc ở mức độ dipeptide, có thể đi qua màng và sau đó được tiêu hóa thành axit amin.
Tiêu hóa các chất nội bào hoặc autophagy
Nhiều thành phần tế bào, chẳng hạn như ty thể, liên tục được loại bỏ khỏi tế bào bởi hệ thống lysosomal. Các bào quan tế bào chất được bao quanh bởi màng của mạng lưới nội chất trơn, tạo thành không bào, sau đó các enzyme lysosomal được thải ra trong không bào autophagic và các bào quan được tiêu hóa.
Autophagy là một tài sản chung của các tế bào nhân chuẩn. Những điều này có liên quan đến việc đổi mới các thành phần tế bào.
Sự tiêu hóa của ty thể hoặc các cấu trúc tế bào khác cung cấp một nguồn năng lượng cho các tế bào này. Sau khi tiêu hóa cấu trúc tế bào, không bào autophagic có thể trở thành cơ thể còn sót lại.
Họ có vai trò trong sự biến thái
Gần đây, vai trò của lysosome đã được phát hiện trong sự biến thái của ếch. Sự biến mất của đuôi ấu trùng của con nòng nọc của ếch là do hoạt động lysosomal (hành động của các cathepsin có trong lysosome).

Quá trình biến thái của ếch có sự góp mặt của lysosome
Chúng giúp tổng hợp protein
Các nhà khoa học Novikoff và Essner (1960) đã đề xuất vai trò có thể có của lysosome trong quá trình tổng hợp protein. Ở gan và tuyến tụy của một số loài chim, lysosome dường như hoạt động mạnh hơn và phát triển cho thấy đây là mối quan hệ có thể với chuyển hóa tế bào.
Họ giúp thụ tinh
Trong quá trình thụ tinh, đầu của tinh trùng tiết ra một số enzyme lysosomal giúp cho sự xâm nhập của tinh trùng trong lớp vitelline của noãn.
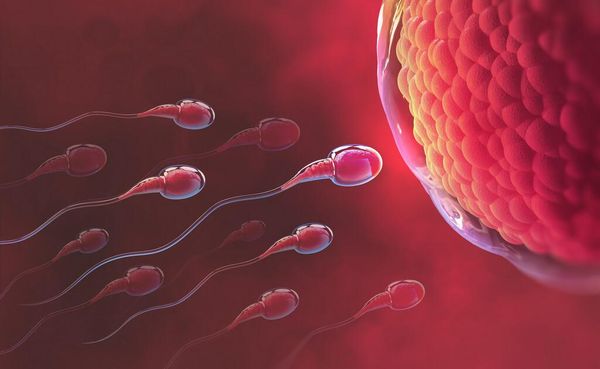
Đầu tinh tùng sẽ tiết ra enzyme lysosomal hỗ trợ xâm lấn vào noãn của trứng
Các acrosome chứa protease và hyaluronidase và axit phosphatase dồi dào. Hyaluronidase phân tán trong các tế bào xung quanh tế bào trứng và protease tiêu hóa zona pellucida tạo ra một kênh thông qua đó nhân tinh trùng xâm nhập.
Nó có vai trò trong sự tạo xương
Người ta đã lập luận rằng sự hình thành của các tế bào xương và sự phá hủy của chúng phụ thuộc vào hoạt động lysosomal. Tương tự, sự lão hóa tế bào và sự phát triển parthenogenetic có liên quan đến hoạt động của lysosome.
Osteoclasts (tế bào đa nhân) loại bỏ xương, làm như vậy bằng cách giải phóng các enzyme lysosomal làm suy giảm ma trận hữu cơ. Quá trình này được kích hoạt bởi hormone tuyến cận giáp.
Tự động hóa sụn và mô xương
Vitamin A dư thừa gây ngộ độc tế bào. Nó làm gián đoạn màng lysosomal, gây ra sự giải phóng các enzyme trong tế bào và gây ra sự tự phân hủy trong sụn và mô xương.
Các bệnh do rối loạn lysosome
Các bệnh gây ra do rối loạn Lysome thường bắt nguồn từ sự thiếu hụt hoặc rối loạn một hay nhiều loại enzyme nào đó, gây ứ động các chất thải, làm chết tế bào, một lượng lớn tế bào thoái hóa hoặc chết đi có thể gây ra bệnh lý vô cùng nguy hiểm, có thể kể đến ung thư trong trường hợp này. Thường xảy ra do rối loạn gen mã hóa của các enzyme này.
Tuy nhiên một số protein liên quan đến quá trình chuyển hóa và phóng thích các chất trong Lysosome cũng là căn nguyên của các chứng rối loạn Lysosome.
Bệnh do rối loạn Lysosome rất đa dạng, triệu chứng phát bệnh không rõ ràng thường là mất điều hòa nhiệt độ cơ thể, co giật, cứng khớp không rõ nguyên nhân, đau xương không rõ nguyên nhân, đục giác mạc, phù thai nhi v.v… Mà tính chất của bệnh rất nặng, giai đoạn biểu hiện lâm sang ra bênh ngoài là giai đoạn cuối, quá trình điều trị sẽ rất phức tạp. Vì thế nên có cách phòng tránh ngay từ ban đầu.
Bệnh lysosomal
Bệnh Gaucher loại I, II và III
Bệnh Gaucher là loại rối loạn lưu trữ lysosomal phổ biến nhất. Các nhà nghiên cứu đã xác định ba loại bệnh Gaucher khác nhau dựa trên sự vắng mặt (loại I) hoặc sự hiện diện và mức độ của (loại II và III) biến chứng thần kinh.
Hầu hết những người bị ảnh hưởng đều có loại I, có thể bị bầm tím, mệt mỏi mãn tính và gan to và / hoặc lách to bất thường (hepatosplenomegaly).

Trẻ em là đối tượng dễ mắc bệnh Gaucher nhất
Bệnh Gaucher loại II xảy ra ở trẻ sơ sinh và trẻ sơ sinh, và được đặc trưng bởi các biến chứng thần kinh có thể bao gồm co thắt cơ bắp không tự nguyện, khó nuốt và mất các kỹ năng vận động có được trước đó..
Bệnh Gaucher loại III xuất hiện trong thập kỷ đầu tiên của cuộc đời. Biến chứng thần kinh có thể bao gồm suy thoái tinh thần, không có khả năng phối hợp các cử động tự nguyện và co thắt cơ bắp của cánh tay, chân hoặc toàn bộ cơ thể.
Các loại bệnh Niemann-Pick A / B, C1 và C2
Bệnh Niemann-Pick bao gồm một nhóm các rối loạn di truyền liên quan đến quá trình chuyển hóa chất béo. Một số đặc điểm chung cho tất cả các loại bao gồm mở rộng gan và lá lách. Trẻ em mắc bệnh Niemann-Pick, loại A hoặc C, cũng bị mất dần các kỹ năng vận động, khó khăn trong việc ăn uống, khó khăn trong học tập tiến triển và co giật.
Bệnh Fabry
Các triệu chứng của bệnh Fabry thường bắt đầu trong thời thơ ấu hoặc thanh thiếu niên, nhưng có thể không rõ ràng cho đến thập kỷ thứ hai hoặc thứ ba của cuộc đời.
Bệnh Fabry là một bệnh dễ gặp với những đối tượng mà lysosome bị rối loạn
Các triệu chứng đầu tiên bao gồm các cơn đau rát nghiêm trọng ở tay và chân. Các dấu hiệu ban đầu khác có thể bao gồm giảm sản xuất mồ hôi, khó chịu khi gặp nhiệt độ ấm áp và sự xuất hiện của phát ban da màu đỏ đến xanh đậm, đặc biệt là ở khu vực giữa hông và đầu gối..
Bệnh lưu trữ Glycogen II (bệnh Pompe)
Bệnh của Pompe có dạng khởi phát muộn. Bệnh nhân có dạng trẻ sơ sinh bị ảnh hưởng nặng nề nhất. Mặc dù những đứa trẻ này thường xuất hiện bình thường khi sinh, bệnh xảy ra trong vòng hai đến ba tháng đầu với tình trạng yếu cơ tiến triển nhanh, giảm trương lực cơ (hạ huyết áp) và một loại bệnh tim gọi là bệnh cơ tim phì đại..
Vấn đề cho ăn và khó thở là phổ biến. Dạng người chưa thành niên / người trưởng thành xảy ra giữa thập kỷ thứ nhất và thứ bảy do yếu cơ dần dần hoặc có triệu chứng suy hô hấp.
Gangliosidosis loại I (bệnh Tây Sachs)
Có hai dạng bệnh Tây Sachs chính: dạng cổ điển hoặc trẻ sơ sinh và dạng khởi phát muộn.
Ở những người mắc bệnh thời thơ ấu của Tay Sachs, các triệu chứng thường xuất hiện lần đầu tiên từ ba đến năm tháng tuổi. Chúng có thể bao gồm các vấn đề về ăn uống, điểm yếu chung (thờ ơ) và phản xạ giật mình quá mức để đáp ứng với tiếng ồn lớn và đột ngột. Sự chậm trễ của động cơ và suy giảm tinh thần là tiến bộ.

Bệnh Tây-sachs khiến cho các hoạt động thường nhật của trẻ bị ảnh hưởng nghiêm trọng
Ở những người có dạng khởi phát muộn, các triệu chứng có thể xuất hiện bất cứ lúc nào từ tuổi thiếu niên đến 30 tuổi. Hình thức trẻ sơ sinh thường tiến triển nhanh chóng, dẫn đến suy giảm đáng kể về tinh thần và thể chất.
Một triệu chứng đặc trưng của bệnh Tay Sachs, xảy ra trong 90 phần trăm các trường hợp, là sự phát triển của các đốm đỏ ở phía sau mắt. Các triệu chứng của bệnh Tây Sachs khởi phát khác nhau tùy theo từng trường hợp. Rối loạn này tiến triển chậm hơn nhiều so với dạng trẻ sơ sinh.
Gangliosidosis loại II (bệnh Sandhoff)
Các triệu chứng đầu tiên của bệnh Sandhoff thường bắt đầu từ ba đến sáu tháng tuổi. Bệnh không thể phân biệt được về mặt lâm sàng với bệnh gangliosidosis loại I.
Bệnh bạch cầu đa nhân
Các dấu hiệu và triệu chứng đầu tiên có thể mơ hồ và dần dần, vì vậy rối loạn này rất khó chẩn đoán. Đi bộ không ổn định thường là triệu chứng quan sát đầu tiên.
Đôi khi, triệu chứng sớm nhất là sự chậm phát triển hoặc suy giảm thành tích học tập. Theo thời gian, các triệu chứng có thể bao gồm co cứng rõ rệt, co giật và chậm phát triển tâm thần sâu sắc.
Bệnh lưu trữ của mucopolysacarit (bệnh Hurler và các biến thể, loại A, B, C, D, Morquio Loại A và B, bệnh Maroteaux-Lamy và Sly)
Những bệnh này được gây ra bởi sự thay đổi trong sự phân hủy bình thường của carbohydrate phức tạp được gọi là mucopolysacarit. Những bệnh này có một số đặc điểm chung, bao gồm biến dạng xương và khớp gây cản trở khả năng vận động và thường gây ra viêm xương khớp, đặc biệt là các khớp lớn hỗ trợ cân nặng.

Tất cả các bệnh này, ngoại trừ bệnh Sanfilippo, cản trở sự tăng trưởng, gây tầm vóc ngắn.
Bệnh Schindler loại I và II
Bệnh loại I của Schindler là dạng cổ điển, xuất hiện lần đầu tiên trong thời thơ ấu. Các cá nhân bị ảnh hưởng dường như phát triển bình thường cho đến khi họ được một tuổi, khi họ bắt đầu mất các kỹ năng có được trước đó, đòi hỏi sự phối hợp của các hoạt động thể chất và tinh thần.
Schindler Type II là hình thức xuất hiện ở người lớn. Các triệu chứng có thể bao gồm sự phát triển của các cụm đổi màu tương tự như mụn cóc trên da, mở rộng vĩnh viễn các nhóm mạch máu gây đỏ da ở các khu vực bị ảnh hưởng, dày lên các đặc điểm trên khuôn mặt và suy giảm trí tuệ nhẹ.
Bệnh Batten
Bệnh Batten là dạng thiếu niên của một nhóm các rối loạn thần kinh tiến triển được gọi là lipofuscinosis tế bào thần kinh. Nó được đặc trưng bởi sự tích tụ của một chất béo trong não, cũng như trong các mô không chứa các tế bào thần kinh.

Bệnh Batten cũng là một bệnh có liên quan đến sự rối loạn của lysosome
Bệnh Batten được đánh dấu bằng suy giảm thị lực tiến triển nhanh chóng (teo mắt) và rối loạn thần kinh, có thể bắt đầu trước tám tuổi. Nó xảy ra chủ yếu ở các gia đình gốc Scandinavi từ Bắc Âu và rối loạn ảnh hưởng đến não và có thể gây suy giảm trí tuệ và chức năng thần kinh.
Dân số bị ảnh hưởng
Là một nhóm, người ta tin rằng các bệnh lưu trữ lysosomal có tần suất ước tính khoảng một trong 5.000 ca sinh sống. Mặc dù các bệnh riêng lẻ rất hiếm, nhưng toàn bộ nhóm ảnh hưởng đến nhiều người trên thế giới.
Một số bệnh có tỷ lệ mắc cao hơn ở một số dân. Ví dụ, các bệnh của Gaucher và Tay-Sachs thường xuyên hơn trong cộng đồng người Do Thái Ashkenazi. Được biết, một đột biến liên quan đến hội chứng Hurler xảy ra thường xuyên hơn giữa các dân tộc Scandinavi và Nga.
Chẩn đoán
Chẩn đoán trước sinh có thể cho tất cả các rối loạn lưu trữ lysosomal. Phát hiện sớm các bệnh lưu trữ lysosomal, trước khi sinh hoặc càng sớm càng tốt, rất quan trọng bởi vì khi điều trị có sẵn, cho chính bệnh hoặc cho các triệu chứng liên quan, chúng có thể hạn chế đáng kể quá trình dài hạn và tác động của bệnh.
Cách phòng tránh các bệnh về lysosome
Xét nghiệm tổng quát định kỳ
Có thể theo dõi tình trạng hoạt động của Lysosome thông qua xét nghiệm nước tiểu, máu, phân tích nguyên bào sợi da… nên quá trình phòng bệnh điều trị bệnh sẽ đơn giản hơn.
Hạn chế thức ăn dầu mỡ, rượu bia thuốc lá
Căn nguyên của tất cả các chứng bệnh đều đến từ thói quen sống không lành mạnh, vai trò của Lysosome tiêu hóa sản phẩm thừa, khi tế bào tiếp nhận quá nhiều chất độc hại áp lực hoạt động của Lysosome sẽ tăng, như một bộ máy quá tải, hiệu suất hoạt động sẽ giảm, tế bào sẽ phải chết khi ứ động quá nhiều sản phẩm thừa.

Bia rượu và các chất kích thích là nguyên nhân dẫn tới các rối loạn lysosome
Có biện pháp trung hòa lại các gốc tự do tấn công từ môi trường
Gốc tự do cũng là một tác nhân giết chết tế bào, gây ra các bệnh mãn tính nguy hiểm như ung thư, tim mạch, tiểu đường v.v… gốc tự do đến từ môi trường sống ô nhiễm, thực phẩm mang tính axit cao, hằng ngày cơ thể đối mặt với hơn 10.000 gốc tự do, nên có biện pháp bảo vệ mình hiệu quả
Ngay cả trường hợp rối loạn Lysosome do di truyền cũng có thể điều trị nếu phát hiện sớm và có cách phòng tránh đúng cách. Cuộc sống và sức khỏe nằm trong tay chúng ta, hành động thế nào mới là điều đáng quan tâm.
Hy vọng bài viết trên đã giúp mọi người hiểu thêm về lysosome là gì và chức năng tuyệt vời của lysosome, từ đó biết được các bệnh liên qua để có cách kiểm tra, phát hiện và điều trị kịp thời nếu có các dấu hiệu nghi ngờ là bệnh lý rối loạn lysosome. Chúc các bạn luôn khỏe mạnh!
Xem các bài viết liên quan:


